






INTRODUCTION
Duck consumption in Korea has steadily increased over the years, from 0.97 kg in 2005 to 2.93 kg in 2020 and 3.65 kg in 2023. The native duck industry is an important sector in Korea. Korean native ducks, developed as a pure breed by the National Institute of Animal Science (NIAS), originated from wild migratory ducks that were initially domesticated and later bred into broiler ducks (Cho et al., 2023). Although Korean native ducks exhibit lower growth rates and productivity compared to imported broiler ducks, they are rich in beneficial fatty acids and are highly preferred by consumers due to their tender texture and higher shear force (Kwon et al., 2014).
In the duck industry, understanding the genetic structure and diversity of duck populations plays a crucial role in meeting consumer demands and improving production efficiency. Analyzing genetic diversity and population structure provides valuable foundational data for species conservation and breeding programs (Gajaweera et al., 2019). Moreover, Korean native ducks exhibit two primary plumage types, colored and white, which may be associated with distinct genetic structures and characteristics. In poultry, feather coloration is a significant trait linked to aesthetic, physiological, and industrial importance. Thus, it is necessary to classify native duck populations based on feather color and investigate their genetic structure and diversity to support sustainable breeding practices (Sultana et al., 2018).
In this study, the genetic structure and diversity of Korean native ducks with colored and white plumage phenotypes were analyzed using 30K SNP chip data. The aim of this research is to compare the genetic structures of colored and white feathered duck populations, identify their genetic diversity, and provide insights for preserving the genetic resources of Korean native ducks.
MATERIALS AND METHODS
The animal care and experimental procedures in this study were conducted in accordance with the regulations and approval of the Animal Care and Use Committee of the Poultry Research Institute, National Institute of Animal Science (NIAS).
The Korean native ducks used in this study are domesticated breeds that were developed through hybridization between domestic ducks traditionally raised in Korea and wild mallards, which adapted to the local environment. The National Institute of Animal Science (NIAS) established a stable color line from native ducks collected from private farms. During this process, individuals with white plumage were selectively bred to establish a stable white line. A total of 300 native ducks (150 colored and 150 white) were selected randomly for genotyping using the 30K SNP chip data.
To extract DNA, 2 μL of duck blood was quantified and mixed with lysis buffer, followed by overnight incubation at 65°C in a shaking incubator for nucleic acid solubilization. Automated genomic DNA extraction was performed using the KingFisher system (Thermo Fisher Scientific, Waltham, MA, USA). The extracted DNA was quantified and quality-checked using the EPOCH system (BioTech Inst. Inc., USA) and stored at −20°C.
Genotyping for 300 validated samples was conducted using the newly developed Korean Poultry SNP panel. The Infinium HD Assay Ultra Protocol was applied using Illumina iScan (Illumina, SD, USA) with 200 ng of genomic DNA per sample. Genotype call plots were validated using GenomeStudio software (Illumina, SD, USA), and genotype data were exported in PED and MAP file formats via the Plink plugin for further analysis.
A total of 33,933 SNPs were utilized in this study, with a genotyping call rate of approximately 98.7% for the 300 native duck samples. SNPs with more than 10% missing genotypes (geno>10%) or a minor allele frequency (MAF) below 0.1% were excluded from the analysis. This filtering process resulted in the removal of 576 SNPs due to missing genotypes and 820 SNPs due to low MAF. Consequently, a total of 32,537 high-quality SNPs were retained and used for principal component analysis (PCA). A principal component analysis (PCA) was performed with PLINK1.9 software. PCA was conducted to evaluate breed relationships by utilizing allele frequency data. This multivariate technique reduces the dimensionality of the dataset by summarizing information from a large number of alleles and loci into a smaller number of synthetic variables, referred to as principal components (PCs). These PCs capture the majority of the genetic variation, enabling efficient visualization and interpretation of population structure and genetic relationships among breeds.
The extent of linkage disequilibrium (LD) between markers was quantified using the squared correlation coefficient (r2) of allele frequencies between pairs of loci within an inter-SNP distance of 1 Mb. This analysis was performed both within individual breeds and across all breeds. Pairwise LD between adjacent SNPs was calculated for each chromosome using the default settings in PLINK version 1.9. In this study, the r2 was estimated using the following formula (Dh et al., 2017):
Effective population size (Ne) was estimated based on LD values (r2), and heterozygosity over the next 50 generations was projected using statistical analyses in R software. In this study, the effective population size (Ne) was estimated using the following formula (Edea et al., 2013):
The effective population size (Ne) at a given generation (t) was estimated using the following formula:
, where C represents the genetic distance between markers, expressed in Morgans. This formula assumes that linkage disequilibrium (r2) between closely located markers reflects recombination events occurring in more recent generations, while r2 between more distant markers represents older historical recombination (Dh et al., 2017).
RESULTS
The analysis of genetic diversity and population structure in native duck breeds provides essential insights for species conservation and breeding improvement. Understanding the genetic variation within and between native duck populations allows the identification of unique genetic traits associated with adaptation, resilience, and productivity. These findings are crucial for developing effective conservation strategies and breeding programs aimed at enhancing the genetic diversity and efficiency of genomic selection of native duck populations (Jin et al., 2023).
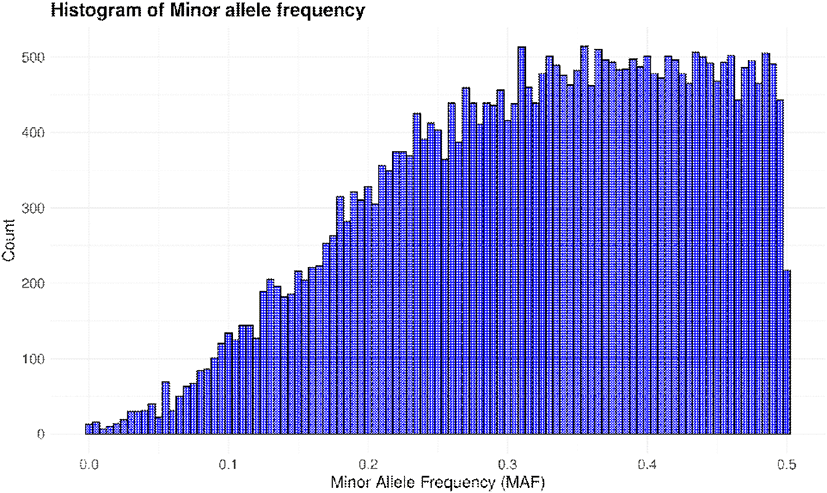
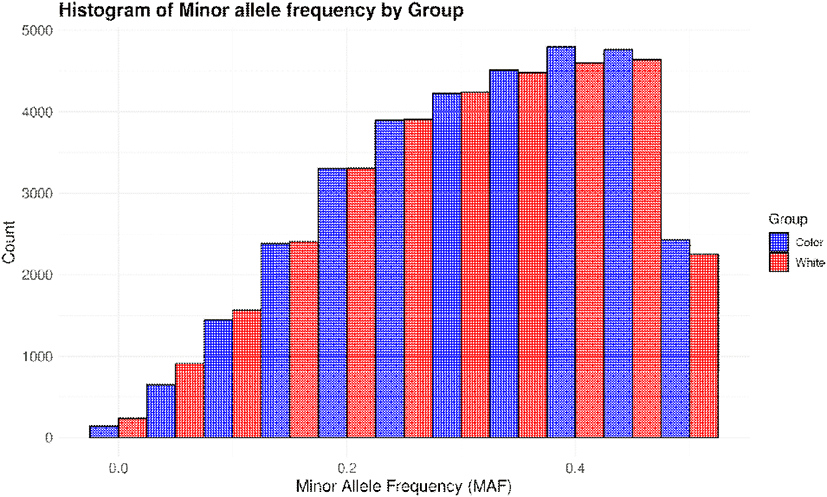
Before proceeding with the analysis, we first examined the MAF frequency distribution and genotype quality for the 300 native ducks included in this study. A histogram of MAF was generated for the entire population of 300 ducks to evaluate allele frequency patterns. Additionally, the dataset was divided into colored and white groups, and the differences in MAF distributions between the two groups were calculated and visualized (Figs. 1 and 2). The majority of alleles exhibited a tendency for higher frequencies as the MAF approached 0.5. This trend was observed not only in the entire population but also when analyzed separately for the colored and white groups. Additionally, the colored group displayed a higher frequency of alleles with elevated MAF compared to the white group. Moreover, when the MAF exceeded 0.48, a sharp decline in allele frequency was observed across all groups, with the number of alleles dropping to approximately 2,000 (Figs. 1 and 2) (Corredor et al., 2023).
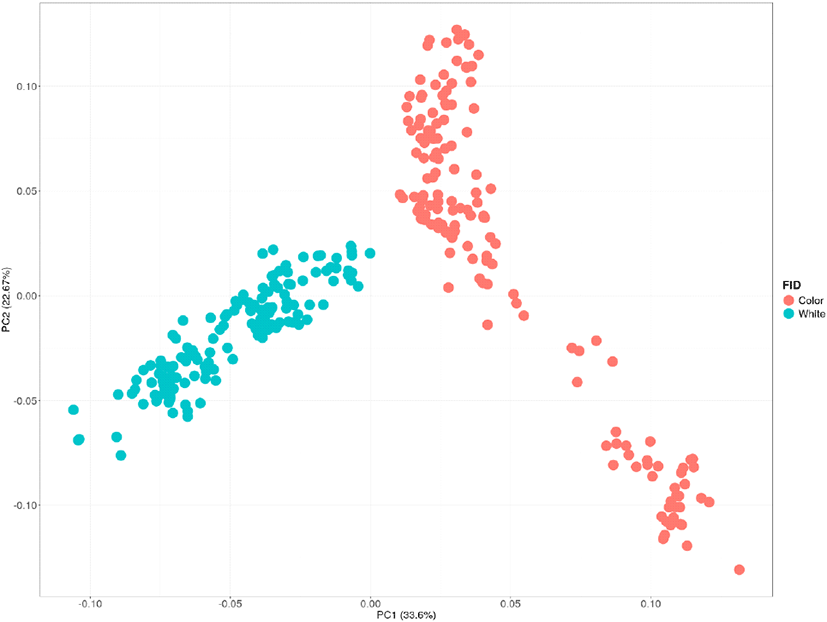
To determine whether the colored and white native duck populations could be genetically distinguished, the Principal Component Analysis (PCA) was conducted. As shown in Fig. 3, red represents the colored group, and blue represents the white group. The analysis revealed that the two groups were clearly separated genetically along Principal Component 1 (33.6%). Additionally, Principal Component 2 (22.67%) indicated that the colored group exhibited greater genetic diversity compared to the white group (Feng et al., 2021). The PCA analysis results showed that the two groups were distinctly clustered, with Principal Components 1 and 2 collectively explaining 56.27% of the total genetic variance. This demonstrates that these two components effectively capture the majority of the genetic differentiation between the colored and white native duck populations (Fig. 3) (Jiang et al., 2021).
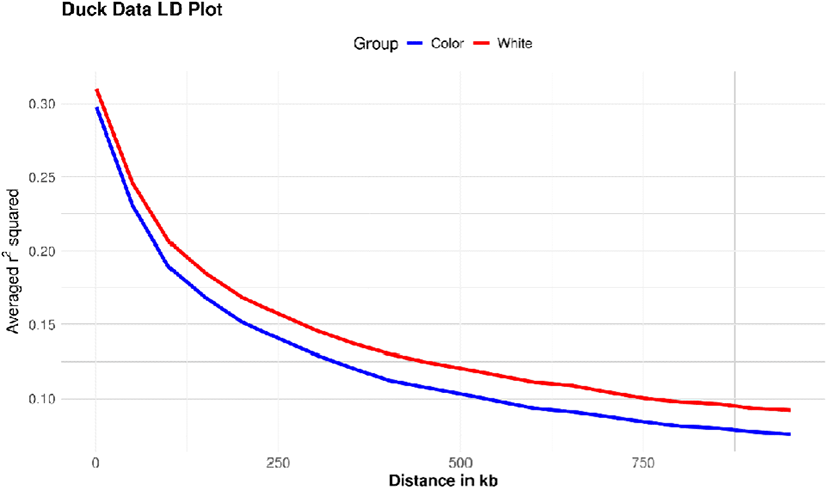
The extent of linkage disequilibrium (LD) in colored and white duck populations is a critical factor in assessing genetic diversity and population structure. The colored group exhibited a faster LD decay compared to the white group, with r2 values sharply decreasing to below 0.2 within 100 kb. In contrast, the white group showed slower LD decay, which may indicate reduced genetic diversity or the presence of a population bottleneck (Fig. 4) (Lin et al., 2022).
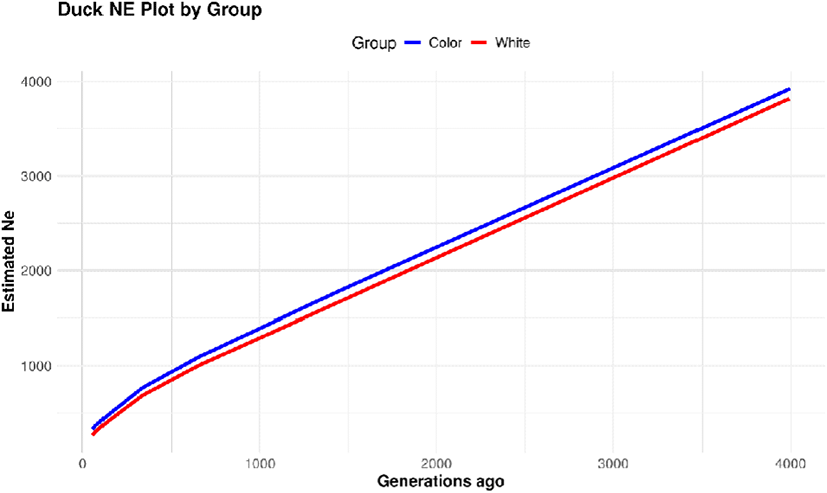
Using the LD information analyzed for each group, the effective population size (Ne) was estimated. The effective population size is defined as “the minimum number of individuals required to maintain allele frequencies in a population over successive generations without significant changes”. Thus, Ne is a critical indicator for managing inbreeding and ensuring sustainable population improvement (Fig. 5) (Edea et al., 2013). The Ne estimation revealed that the white group had a smaller effective population size compared to the colored group. Over 50 generations, the Ne was estimated to be approximately 320 for the colored group and 260 for the white group. According to the International Union for Conservation of Nature (IUCN.org), populations with a Ne of fewer than 50 individuals are considered critically endangered. The average Ne for both colored and white native duck populations was approximately 290, indicating that these populations are currently maintaining a sustainable effective population size (Garner et al., 2020).
DISCUSSION
This study provides comprehensive insights into the genetic diversity and population structure of colored and white native duck populations, highlighting critical differences and implications for conservation and breeding programs. Principal Component Analysis (PCA) clearly distinguished the colored and white groups along the first principal component (PC1), which accounted for 33.6% of the total variance. This genetic separation underscores significant population differentiation between the two groups, likely driven by their distinct breeding histories and selective pressures. Furthermore, the greater genetic diversity observed in the colored group, as indicated by PC2 (22.67%), suggests that this group has maintained a more diverse gene pool compared to the white group. The white group was established by selecting individuals with white plumage from the colored ducks (Lim et al., 2024). The phenotype-based selection process within the colored group is presumed to have led to reduced genetic diversity in the white duck group compared to the colored ducks. The genetic pool differences between the colored and white ducks observed in PCA are likely attributed to allelic differences affecting plumage color. To confirm these findings, further studies are required, including GWAS and selection signature analyses, to identify the causal variants. In conclusion, the findings emphasize the need for targeted conservation strategies and breeding programs tailored to the specific genetic characteristics of each population. The higher genetic diversity and faster LD in the colored group highlight its potential as a genetic resource for sustainable breeding. In contrast, the white group’s lower genetic diversity and smaller Ne underscore the importance of mitigating inbreeding and maintaining a viable population size to ensure its long-term genetic health. Future studies should focus on identifying feather color-associated genes and exploring their functional implications to further enhance the understanding and management of native duck populations. For example, identifying genes or markers associated with feather color and applying marker-assisted selection (MAS) based on these findings could enable the establishment of duck populations with consumer-preferred feather colors. This research would have direct industrial applications, contributing significantly to the improvement and sustainability of the duck industry.
SUMMARY
This study was conducted to analyze the genetic diversity and structure of native duck populations with colored and white plumage phenotypes using 30K SNP chip data. For the study, 300 native duck samples (150 colored and 150 white) were provided by the Poultry Research Institute of the National Institute of Animal Science. The results showed that the Minor Allele Frequency (MAF) between the two groups was similar, but the overall frequency sharply declined when the MAF was above 0.48, with only about 2,000 SNPs detected. Principal Component Analysis (PCA) based on the identified SNPs revealed that the colored and white groups were clearly separated along Principal Component 1 (33.6%). Furthermore, genetic diversity inferred from Linkage Disequilibrium (LD) and Effective Population Size (Ne) indicated that the colored group exhibited slightly greater genetic diversity than the white group. These findings confirm that there are genetic structural differences between the two populations. Further analysis is necessary to identify genes associated with plumage color in native ducks.