






INTRODUCTION
The introduction of next-generation sequencing (NGS) technology in the early 2000s brought about dramatic changes in the field of biotechnology research (Mardis, 2013). This tendency led to a decrease in analysis cost and an increase in accuracy over time, and as a result, it provides a possibility of genome analysis for large-scale populations (Hu et al., 2021). In particular, NGS technology has been utilized in various species for de novo genome sequencing and DNA resequencing (Park and Kim, 2016). Also, the accumulated sequences are applied to identify the genetic markers such as single nucleotide polymorphism (SNP) and insertion and deletion (InDel).
Ducks are one of the most useful protein sources in the poultry industry, following chickens. The breast meat of ducks is close to red unlike chicken, so it has similar sensory characteristics to beef and pork (Chartrin et al., 2006). Also, it has a higher unsaturated fatty acid content than other meats, so consumer interest is continuously increasing (Onk et al., 2019).
The Korean native ducks (KNDs) are a pure breed that was bred from wild migratory ducks by the National Institute of Animal Science (NIAS) in Korea to be used in duck farming, then improved to be used as broiler ducks. Although it has a lower growth rate and productivity than imported broiler ducks, it has a high content of useful fatty acids. Also, the tenderness and shearing power of the meat are high, so it has a high preference of consumers (Hong et al., 2012; Kim et al., 2012). However, in the case of native ducks, the most of plumage color in the group is almost fixed to black-brown, so there are limitations to their use as meat, and in the end, the Pekin species imported from overseas accounts for 90% of the domestic broiler duck market. Therefore, to improve the market share of KNDs for broiler use, it is necessary to understand the genetic background of white KNDs and utilize this to introduce genetic improvement and selection systems.
Based on this background and necessity, this study was conducted to identify the genomic information of KNDs using genomic data of colored and white KNDs and search for the candidate genes associated with plumage color.
MATERIALS AND METHODS
The KND samples were provided by the NIAS in Korea. The KND population consisted of two groups: colored KND (n = 2) and white KND (n = 2). The blood samples were collected from the brachial vein and used for extraction of genomic DNA (gDNA). gDNA was extracted using the PrimePrep Genomic DNA Extraction Kit from Blood (GeNetBio, Daejeon, Korea), following the manufacturer’s instructions. The purity and concentration of the extracted gDNA were confirmed using a NanoDrop 2000c spectrophotometer (Thermo Fisher Scientific, Waltham, MA, USA). The final gDNA was stored at −20°C until use.
The gDNA library was constructed according to the NGS library preparation workflow (TruSeq Nano DNA library prep; Illumina, San Diego, CA, USA), and the raw sequence data were acquired through Illumina NovaSeq 6000 platform (Illumina, San Diego, CA, USA), resulting in the generation of 151 bp paired-end reads. The raw sequence reads were pre-processed following the quality control (QC) procedure to remove unusable reads. The sequences with less than 10% of their total lengths were removed.
The procedure of calling variants from raw sequence data was performed in accordance with the GATK best practice workflow. All clean reads were aligned to the chromosome-level reference genome of the Anas platyrhynchos (assembly version ZJU1.0; accession number GCF_015476345.1) downloaded from the NCBI RefSeq database using the “BWA-MEM” (Li, 2013) with default parameters. Following the alignment step, the SAM format data were converted to BAM format and indexed by the “SAMtools” (Li et al., 2009), and duplicated reads were filtered using the “MarkDuplicates” program in the “GATK” tool (version 4.1.4.1) (McKenna et al., 2010). Since no available variant information was provided for the present version of the reference genome, the recalibration step was performed slightly differently from the existing method. The first variant calling step was performed using the “HaplotypeCaller”, and the output variants were filtered as following criteria: QD<2.0, FS>60.0, MQ<40.0, MQRankSum<−12.5, ReadPosRankSum<−8.0. Then, the recalibration step was performed using the filtered variants, and the second variant calling step proceeded with recalibrated data. In the end, the output data was filtered using the same criteria as the previous filtering step.
The variant annotation for the final variants of four KNDs was performed using the “SnpEff” program (Cingolani et al., 2012). After the annotation step, the common differential variants were distinguished to identify the genetic difference in genomes between colored and white KNDs. The workflow was as follows: we first collected the variants with the same genotype in each group and then determined the differential variants between the two groups. Subsequently, these common differential variants were used for functional enrichment analysis. The Gene Ontology (GO) analysis and the Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway analysis for the gene sets containing the common differential variants were conducted using the “g:Profiler” online web server (https://biit.cs.ut.ee/gprofiler/gost) (Raudvere et al., 2019). The conditions for filtering significant analysis results were as follows: significance threshold was P<0.05, minimum term size was 10 and maximum term size was 350.
RESULTS AND DISCUSSION
NGS resequencing was performed on four birds from each KND group. Table 1 summarizes the results of resequencing. The output indicated that more than 132,124,410 of 151 bp paired-end short reads were acquired from each bird, with total sequence lengths exceeding 39 Gb. The sequencing quality was high, with a Q30 ratio of more than 90%. The average sequencing depth of coverage was found to be approximately 35X.
KND, Korean native duck.
A total of 3,713,890 SNPs and 795,089 InDels were identified in the colored KNDs, and a total of 3,682,977 SNPs and 784,006 InDels were identified in the white KNDs. For the common variants between the two groups, a total of 1,887,587 SNPs and 419,135 InDels were identified (Table 2).
SNP, single nucleotide polymorphism.
Table 3 shows the variant effects by functional class as the results of variant annotation using “SnpEff”. More than 16 million effects were identified in the colored and white KNDs, and among them, the effect of mutations common to the two groups was confirmed to be about half. In addition, about 30% of the confirmed variants were found to be non-synonymous mutations (missense and nonsense).
Table 4 indicates the results aggregated by region where the effects of the variants shown in Table 3 were located on the genome. According to these results, the region with the most variants was in the intron, followed by the variants in the upstream, downstream, and intergenic regions. Although there were relatively few variants of more than 2% in the exon region, it was confirmed that some variants with a high impact of the variant effect, such as frameshift or exon loss variants, existed in the exon region.
A total of 1,248,986 variants with the same genotype in the colored KND group were identified, and a total of 1,253,989 variants were identified in the white KND group. Among these variants, 753,175 variants existed in common within the two groups, and ultimately, a total of 210,937 common differential variants were confirmed between the colored and white KNDs. The putative variant impacts of each variant were classified as follows: high (186), moderate (993), low (3,012), and modifier (206,746). These variants participated in 7,983 genes and these genes were used for functional enrichment analysis.
As a result of analysis using 7,982 genes, a total of 337 GO terms (42 molecular function terms, 258 biological process terms, and 37 cellular component terms) and 2 KEGG pathways (Wnt signaling pathway and MAPK signaling pathway) were observed (P<0.05) (Fig. 1A). To further increase the significance of the results, the term size (the number of genes associated with a given GO term) was adjusted between 10 and 350, resulting in a total of four terms in the molecular function category, 14 terms in the biological process category, eight terms in the cellular component category, and one KEGG pathway were significantly identified (Fig. 1B).
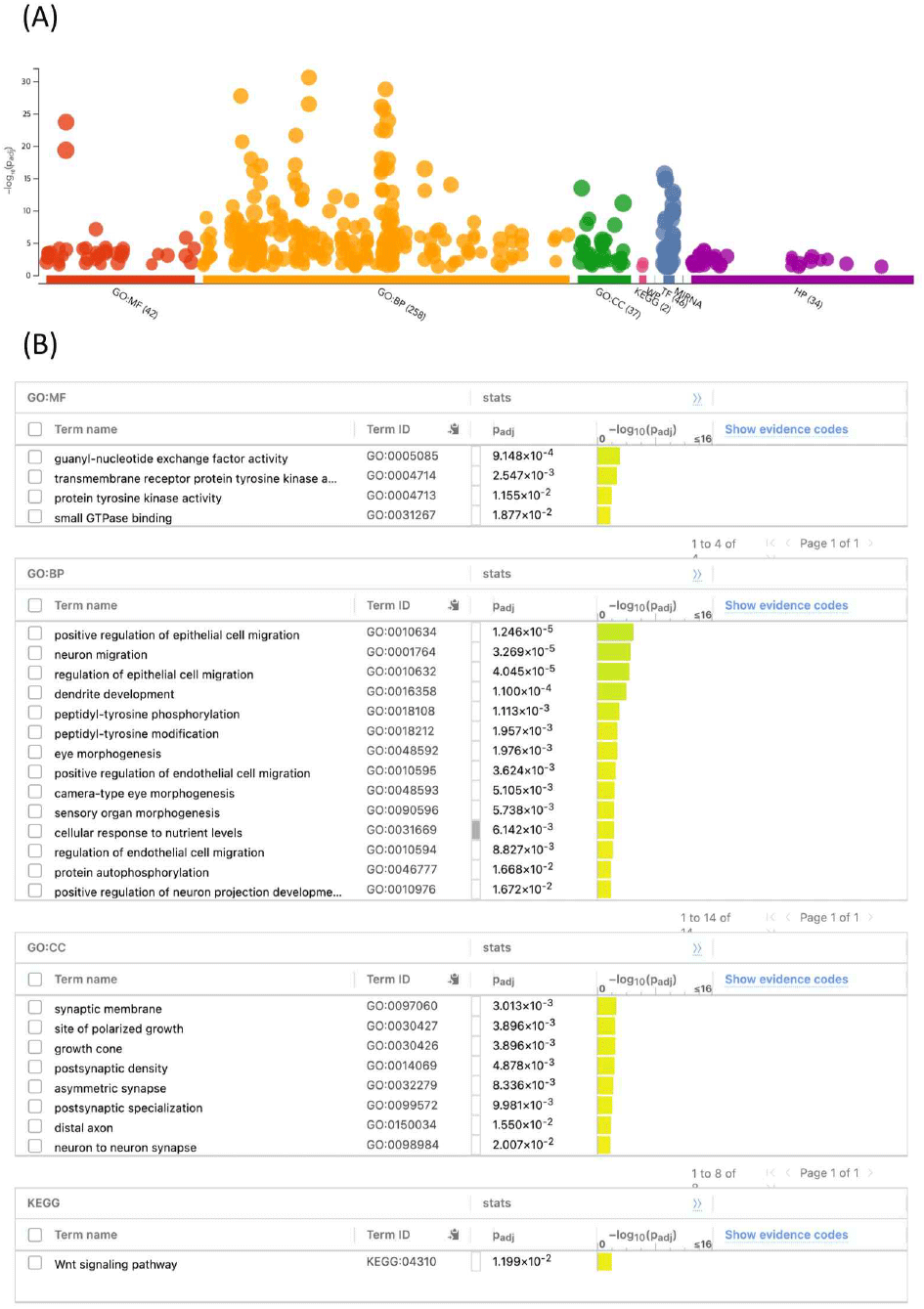
Among the identified GO terms, the significantly related terms with the KND phenotype were as follows: transmembrane receptor protein tyrosine kinase activity (GO: 0004714), protein tyrosine kinase activity (GO:0004713), peptidyl-tyrosine phosphorylation (GO:0018108), and peptidyl-tyrosine modification (GO:0018212). All identified significant GO terms were related to tyrosine activity, and according to previous studies, they have been reported to be related to the mechanism of melanin present in muscles or feathers (Li et al., 2012; Kulikova, 2021; Zhang et al., 2022; Xu et al., 2023). In addition, some of the genes such as EGFR, PDGFRA, and DDR2 included in each term category were confirmed to affect tissue pigmentation during melanin synthesis and regulation (Quillen et al., 2012; Hulsman et al., 2014; Reger de Moura et al., 2020). Therefore, the GO terms and genes presented as a result of this study suggest the possibility and necessity of follow-up research to explain the genetic background of plumage color formation in KND.
SUMMARY
In this study, we used NGS resequencing data of colored and white KND to screen the genetic characteristics of KND by comparing variants present in the two groups. In addition, through the comparison results, the putative candidate gene information that affects the plumage color of KND was confirmed. This result could be significant in understanding the genetic characteristics of KND and establishing a research foundation for the genetic improvement of white KNDs.