






INTRODUCTION
Avian influenza virus (AIV) must utilize host cellular proteins as well as their own proteins to complete their life cycle. For example, AIV utilizes cellular transcriptional machineries such as mini-chromosome maintenance (MCM) complex (Kawaguchi and Nagata, 2007) chromatin remodelers such as chromatin remodeling protein 1 (CHD1), chromatin remodeling protein 6 (CHD6), nuclear matrix protein 2 (NXP2), and pol II function modulator CLE (Alfonso et al., 2011; Rodriguez et al., 2011; Ver et al., 2015; Marcos-Villar et al., 2016) for both viral transcription and replication. Watanabe et al. identified 1,291 host factors that are co-immunoprecipitated with viral protein such as HA, NP, M1, and M2 in Human Embryonic Kidney (HEK) cells using RNAi-based screens (Watanabe et al., 2014) and Karlas et al. discovered 287 human cellular factors that are involved in influenza A virus replication using RNAi-based screens in HEK293T reporter cells (Karlas, 2010).
Recently, mammalian (human, mouse) Fragile X mental retardation protein (FMRP) has been reported to stimulate influenza A virus ribonucleoprotein (vRNP) assembly and help export vRNP from the nucleus to the cytoplasm by binding to vRNP (Zhou, 2014). In addition, human FMRP restricted zika virus by repressing viral protein translation (Zalfa et al., 2003; Soto-Acosta, 2018). Thus, FMRP has been focused as a novel host cellular protein that can be the target for the development of new anti-viral drugs, which have the advantage in that they have wide spectrum effects on many types or subtypes of viruses while anti-viral drugs targeting viral proteins are frequently dismayed when new types of viruses emerge due to genetic reassortment (Marshall et al., 2013; van de Wakker et al., 2017; Zhu et al., 2017). FMRP is an RNA binding protein that consists of three RNA binding domains named KH1, KH2 and RGG box (Adinolfi et al., 1999; Darnell et al., 2001; Darnell et al., 2005). In humans, the loss of FMRP translational regulator 1 (FMR1) gene expression results in Fragile X syndrome (FXS), autism whose symptoms are the long face, post-pubertal macroorchism, and hyperextensible joints (Mila, 2017). The functional role of FMR1 gene in the chicken system as a host cellular protein for AIV replication, however, is poorly understood.
In this study, we targeted chicken FMR1 (cFMR1) gene using the clustered regularly interspaced short palindromic repeats (CRISPR)-associated protein 9 (CRISPR/Cas9)-mediated genome editing system and overexpressed cFMR1 gene in cFMR1 knockout DF1 cells to examine cFMR1 function as an AIV host factor in the chicken system. Our study will provide insight whether cFMR1 is suitable target gene to generate AIV-resistant drug or animal and emphasize the importance to validate the functional difference of the host factors studied in mammals and birds.
MATERIALS AND METHODS
The CRISPR/Cas9 vector targeting chicken FMR1 (cFMR1) gene (NCBI gene ID: 422395) was constructed using the PX459 vector (Addgene Kit #1000000054, MA, USA) as previously reported (Lee et al., 2017). To insert guide RNA (gRNA) sequence into CRISPR/Cas9 vector, we synthesized sense and antisense oligonucleotides (Table 1) (Bionics, Seoul, Korea) and carried out annealing using the following thermocycling conditions: 30 s at 95°C, 2 min at 72°C, 2 min at 37°C, and 2min at 25°C. The annealed oligonucleotides for each gRNA were ligated into the PX459 vector via the Golden Gate assembly method, and the constructed CRISPR/Cas9 vectors were validated via Sanger sequencing.
The PiggyBac plasmid (Addgene #92078) containing enhanced green fluorescent protein (PiggyBac-eGFP) was digested with AgeI and BsrGI enzymes (New England Biolabs; NEB, UK) to create a linearized vector. A synthetic protein coding region of chicken FMR1 (NCBI gene ID: 422395) and human FMR1 (NCBI gene ID: 2332) (Bionics) was cloned into the linearized vector using Takara In-Fusion Ligation mix (Takara, Kasatsu, Japan) according to the manufacture’s protocol. The resulting plasmid was amplified and purified using a Plasmid Maxi Kit (Qiagen, Hilden, Germany). The correct insert was confirmed by sequencing analysis.
The PR8-H5N8 low pathogenic AIVs were generated via reverse genetic systems from eight bidirectional PHW2000 plasmids (a kind gift from Prof. Chang Seon Song of Konkuk University, South Korea) (Chungu et al., 2021) encoding the PB1, PB2, PA, HA, NA, NP, NS, and M genes. Viruses were rescued by co-transfection of the eight bidirectional plasmids into co-cultured Madin-Darby Canine Kidney cells (MDCK; ATCC, VA, USA, #CCL-34) and human 293T embryonic kidney cells (HEK293T; ATCC, #CRL-11268). The generated viruses were grown in MDCK infection medium, consisting of Dulbecco’s minimum essential medium (DMEM; Hyclone, UT, USA) supplemented with 0.3% bovine serum albumin (BSA) (Sigma-Aldrich, MO, USA), 1 × antibiotic-antimycotic reagents (Thermo Fisher-Invitrogen, CA, USA), and 1 µg/mL TPCK-treated trypsin (Sigma-Aldrich), and then incubated at 37°C for 48 hours. The virus stocks were further propagated in 10-day-old embryonated chicken eggs. Aliquots of infectious virus were stored at −80°C for further experiments. All works with low pathogenicity viruses were conducted in a biosafety level 2 facility approved by the Institutional Biosafety Committee, Seoul National University (approval number: SNUIBC-P180625-1).
The viral titration was performed in MDCK cells to determine the median tissue culture infectious dose (TCID50). In brief, viral supernatants of infected cells were used to infect confluent layers of MDCK cells in 96-well plates. Serial dilutions of the supernatant were added to five wells of a 96- well culture plate in triplicate. After 72 to 96 hours, the cytopathic effects (CPEs) were observed and quantified via crystal violet (Sigma-Aldrich) staining. The TCID50 values per mL were calculated using the Spearman-Karber formula (Gilles, 1974).
The chicken DF1 fibroblast cells (ATCC, #CRL-12203) were maintained in DMEM, supplemented with 10% fetal bovine serum (FBS; Hyclone) and 1 × antibiotic-antimycotic reagents at 37°C in an incubator with an atmosphere of 5% CO2 and 60%-70% relative humidity. For transfection of DF1 cells, 2 µg of CRISPR/Cas9 vector targeting cFMR1 gene was mixed with 2 µL of Lipofectamine 2000 reagent (Thermo Fisher- Invitrogen) in Opti-MEM (Thermo Fisher Invitrogen), and this mixture was applied to 2 × 105 DF1 cells in 12-wells culture plates. Approximately 24 hours after transfection, puromycin (GIBCO Invitrogen, NY, USA) (1 µg/mL) was added to the culture medium for the selection of transfected DF1 cells. The complete selection period required 3 to 4 days. Puromycin- selected DF1 clones were seeded into individual wells of a 96-wells plate with culture medium. After clonal expansion of cFRM1 disrupted clones, genomic DNA was extracted from the clones for T7E1 assay and sequencing analysis (Fig. 1).
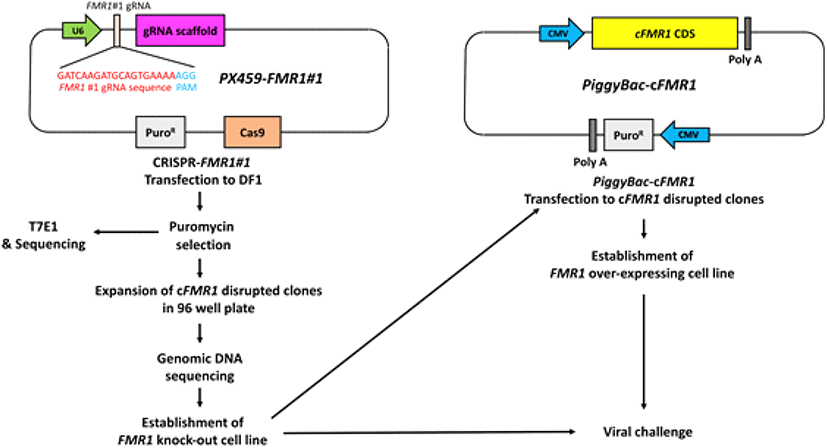
To evaluate the target efficiency of the constructed PX459 vectors in chicken DF1 cells, genomic DNA was extracted from the transfected DF1 cells following puromycin selection. Genomic regions encompassing the CRISPR/Cas9 target sites were amplified using specific primer sets. The primer sets were designed by Primer3Plus online tool (https://3plus.com/cgi-bin/dev/primer3plus.cgi) (Table 1). Following denaturation, the amplicons were reannealed to form heteroduplex DNA. Subsequently, the heteroduplex amplicons were treated with T7 endonuclease I (T7E1, NEB) for 20 min at 37°C and then analyzed via 1% agarose gel electrophoresis. For the sequencing analysis, PCR products containing the target site were cloned into the pGEM-T Easy vector (Promega, WI, USA) and sequenced using an ABI Prism 3730 XL DNA Analyzer (Thermo Fisher-Applied Biosystems, CA, USA) (Fig. 1). The sequences were analyzed against assembled chicken genomes using BLAST (http://blast.ncbi.nlm.nih.gov) and Geneious R6 software (Biomatters Ltd., Auckland, New Zealand).
The total RNA was extracted using a RNeasy mini kit and reverse-transcribed using the Superscript IV First-strand Synthesis System (Thermo Fisher-Invitrogen). The PCR reaction mixture contained 2 μL of PCR buffer, 1 μL of 20 × Eva Green qPCR dye (Biotium, Hayward, CA, USA), 0.5 μL of 10 mM dNTP mixture, 10 pmoles each of target gene- specific forward and reverse primers (Table 1), 1 μL of cDNA, and 1 U of Taq DNA polymerase in a 20 μL final volume. The primer sets were designed by Primer3Plus online tool (https://pirmer3plus.com/cgi-bin/dev/primer3plus.com/cgi-bin/dev/primer3plus.cgi) (Table 1). The RT-qPCR was performed in triplicate. Relative quantification of target gene expression in the infected cells was performed using the following formula: 2-∆∆Ct where ∆∆Ct = (Ct of the target gene - Ct of ACTB) group - (Ct of the target gene - Ct of ACTB) control.
Wild type (WT) DF1 cells and cFMR1 mutated clones were seeded into 24 wells culture plate, and co-transfected after 24 hours with plasmids encoding the PB1, PB2, PA, and NP proteins (200 ng each), together with a plasmid expressing negative-sense luciferase flanked by NS segment noncoding sequences under the control of chicken polymerase I promoter (50 ng), and the 20ng of pGL−4.53 plasmids expressing Renilla luciferase used as the internal reference by using Lipofectamine 2000 transfection reagent. Cells were incubated at 37°C. Twenty hours after transfection, cells were harvested and polymerase activity was measured using Nano-Glo Luciferase Reporter Assay System (Promega).
RESULTS
To disrupt the cFMR1 gene, we used the CRISPR/Cas9 system to introduce a targeted mutation in the cFMR1 gene of chicken DF1 cells. First, we constructed CRISPR/Cas9 vector (hereafter, FMR1#1) targeting exon 9 of the cFMR1 gene to introduce the random indel mutation-mediated premature stop codon (Fig. 2A). We transfected PiggyBac enhanced GFP (eGFP) vector and CRISPR/Cas9 vector in the same manner and used eGFP emission as a transfection efficiency to the DF1 cell. To analyze transfection efficiency, green light emitted from the PiggyBac eGFP transfected DF1 cells was observed (Fig. 2B). After 3∼4 days selection with puromycin, we performed T7E1 assay to investigate genome editing efficiency and the results showed that cleaved bands were detected only in DF1 cells transfected with the CRISPR/Cas9 vector (FMR1#1), indicating that the CRISPR/ Cas9 vectors efficiently induced nucleic acid mutations at the targeted locus of the cFMR1 gene (Fig. 2B). Sequence analysis of the targeted locus using the TA cloning method confirmed the mutations at the targeted regions with 60% (6/10) efficiency (Fig. 2C). Next, we generated cFMR1 mutated clones from DF1 cells transfected with CRISPR/Cas9 targeting cFMR1 (FMR1#1). We generated five cFMR1 gene edited clones from the experimental groups, and sequencing analysis from each clone showed that all of them harbored nucleotide mutations in the cFMR1 gene (genotype: N base pair (bp) deleted (−) or inserted (+) in one allele and M bp deleted (−) or inserted (+) in the other allele is presented ±N bp/±M bp; Clone #1: −12 bp/−12bp, Clone #2:−39 bp/−6 bp, Clone #3: −19 bp/−12 bp, Clone #4 (F112):−35 bp/−9 bp, Clone #5 (F113):−8 bp/+1 bp) (Fig. 2D-E). Among F112 and F113, F113 harbored frameshift mutations (including deleted- or inserted amino acids) at the targeted locus that generated a premature stop codon in the cFMR1 gene. Another clone named F112 harbored deletion at the targeted locus that did not introduce a frameshift. Thus, we selected F113 as a representative clone that harbored disrupted cFMR1 (Fig. 2D-E).
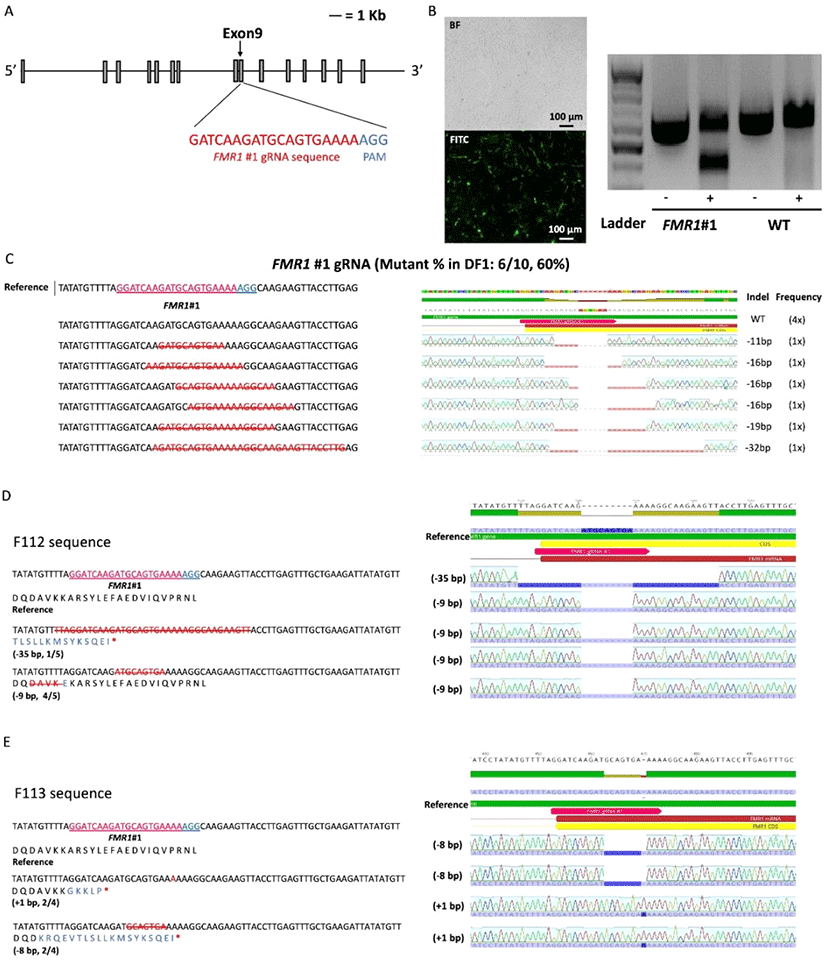
As F112 and F113 harbored different types of indels in both alleles, we analyzed the mRNA sequence of F112 and F113 by sequencing cDNA. While F112 only expressed non-frameshift mutated alleles (Fig. 3A), F113 expressed both of the frameshift mutated alleles (−8 nt deletion allele and +1 nt insertion allele) (Fig. 3B). It was predicted that F112 mRNA did not possess premature stop codon (Fig. 3A) while both alleles of F113 mRNA showed premature stop codon (Fig. 3B). Therefore, we used the F113 as a representative of cFMR1 mutated clone for the subsequent experiment.

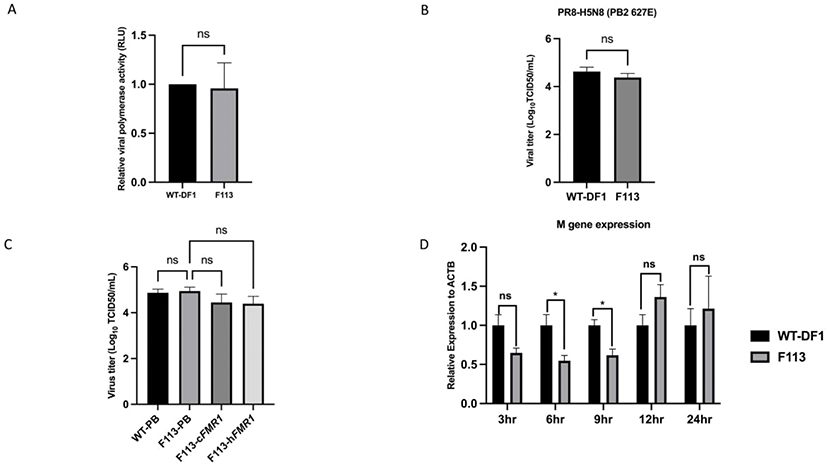
To identify cFMR1 function in AIV, we measured viral polymerase activity via minigenome assay. The viral polymerase activity between WT DF1, and F113 showed non-significant difference at 24 hours post infection (h.p.i) (Fig. 4A). Next, to compare virus progeny generated at 24 h.p.i between WT DF1 and F113, we performed TCID50 assay. The results showed that there was no significant difference of viral titer between WT DF1 and F113 at 24 h.p.i. (Fig. 4B). We also constructed cFMR1 expressing vector (PiggyBac-cFMR1), human FMR1 (hFMR1) expressing vector (PiggyBac-hFMR1) and transfected the vector into F113 to compare the cFMR1 and hFMR1 function in chicken system (Fig. 1). We transfected PiggyBac-eGFP into WT DF1 cells and F113 as a control. Then, we infected WT-PiggyBac eGFP, F113-PiggyBac eGFP, F113- PiggyBac- cFMR1, and F113-PiggyBac-hFMR1 DF1 cells with PR8- H5N8 AIV (Multiplicity of infection, MOI = 0.01). At 24 h.p.i, TCID50 assay was performed and the results showed no significant difference in viral titer among WT-PiggyBac eGFP, F113-PiggyBac eGFP, F113-PiggyBac- cFMR1, and F113-PiggyBac-hFMR1 (Fig. 4C). Finally, to identify whether cFMR1 would function in AIV gene expression in various time points, we infected WT DF1 and F113 with PR8-H5N8 AIV and analyzed viral M gene expression as a representative level of viral gene expression. The viral M gene expression level was significantly decreased in F113 at 6 and 9 h.p.i compared to WT DF1 cells. However, at 12 and 24 h.p.i, the M gene expression level of F113 compared to WT DF1 was not significantly different, which corresponds to minigenome assay, and TCID50 assay results (Fig. 4D).
DISCUSSION
FMRP is dominantly cytoplasmic, however, it has Nuclear Localization Signal (NLS) and Nuclear Export Signal (NES). Therefore, it can shuttle mRNA between nucleus and cytoplasm (Feng et al., 1997; Kim et al., 2009). Also, FMRP is an RNA binding protein, which has three RNA binding domains named KH1 domain, KH2 domain, and RGG box (Adinolfi et al., 1999; Darnell et al., 2001; Darnell et al., 2005). With these properties, FMRP is estimated to associate with 4% of brain mRNA and regulate its transcription (Brown et al., 2001). Due to these features, mammalian (mouse, human) FMRP can bind to NP mediated by vRNA and shuttle vRNP from the nucleus to the cytoplasm.
In this study, we precisely disrupted cFMR1 gene through CRISPR/Cas9 and established cFMR1 mutated clones to investigate its potential role in AIV proliferation. We targeted exon 9 to disrupt KH2 domain of FMRP. The I304N mutation in KH2 domain of human FMRP significantly disrupted the FMRP-NP interaction and reduced the positive effect of FMRP on vRNP assembly (Zhou, 2014). In our results, unlike mammalian FMR1, disruption of KH2 domain coding region in cFMR1 did not affect AIV production at 24 h.p.i. There was no significant difference in viral polymerase activity in cFMR1 disrupted clone (F113) compared to WT DF1. It implies that cFMR1 did not affect viral replication cycle at 24 h.p.i. In addition, there was no significant difference in viral titer, implying that cFMR1 did not affect viral progeny production at 24 h.p.i. To confirm that cFMR1 could not stimulate AIV proliferation, cFMR1 was over-expressed in F113. In addition, hFMR1 was over-expressed in F113 to compare function of cFMR1 and hFMR1 in chicken system. The results showed no significant difference in viral titer among WT-PiggyBac eGFP, F113-PiggyBac eGFP, F113- PiggyBac cFMR1, and F113-PiggyBac hFMR1, confirming that FMR1 (cFMR1, hFMR1) did not affect virus production at 24 h.p.i in chicken cells.
Although cFMR1 did not change virus production at 24 h.p.i, cFMR1 seemed to stimulate proliferation only in an early viral stage. When vRNP is transported into the nucleus at around 1.5 h.p.i, mRNAs are primarily synthesized (transcription) from the parental vRNP. At 3 h.p.i, M gene expression showed no significant difference between WT DF1 and F113, implying that M gene transcript was derived from parental vRNP and similar infection occurred at the initial time. At 6 and 9 h.p.i, however, complementary RNA (cRNA) is synthesized for viral replication (Vreede and Brownlee, 2007; Heldt et al., 2012). At this point, cFMR1 mutation resulted in reduced M gene expression, implying that cFMR1 promoted early viral transcription. During this process, mammalian FMRP is known to help vRNP assembly, and shuttle vRNP from the nucleus to the cytoplasm (Zhou, 2014). Corresponding to previous studies, our results showed significant M gene expression reduction in F113 at 6 and 9 h.p.i., implying that delayed vRNP assembly may result in reduced infectious viral particles and subsequent viral gene expression also reduced.
It is still unknown why cFMR1 only influenced on early stage of AIV life cycle. We speculate that it may be due to either alternative pathway which AIV can exploit or influenza A virus strain/host cell type difference. For example, there is paralog of FMR1 named FMR1 Autosomal Homolog 1 (FXR1) in chicken. FXR1 shares much homology with FMR1, and their protein structure is similar that composes the same domains including RNA binding domains, KH1, KH2, and RGG box (Kirkpatrick et al., 2001). Therefore, AIV may utilize RNA binding property of FXR1 in the absence of FMR1. The exact roles of FXR1 as a host factor for AIV infection remain to be explored in further studies. In addition, A549 cell line (human lung carcinoma cell line) was previously subject to infection with influenza A virus (Zhou, 2014). While A549 cell line was derived from lung which is target of influenza A virus (Zhao et al., 2017), DF1 cells are immortalized chicken fibroblast cell line (Himly et al., 1998) and this discrepancy of cell line may explain modified role of FMRP as a host factor in chicken. The functional comparison of chicken FMRP and FXR1 as an AIV host factor needs to be clarified in further studies.
In conclusion, our findings imply that cFMR1 in DF1 cells does not affect AIV proliferation but stimulate viral gene transcription only at an early stage of the virus life cycle. Therefore, unlike mammalian FMR1, cFMR1 may not be an appropriate candidate for anti-AIV drug development in DF1 cells or AIV resistant chicken line development. Further understanding of cFMR1 in other cell type may provide insight into whether cFMR1 would be an appropriate candidate gene for establishing anti-AIV strategy.